


泛素化看过,连续泛素化看过吗?(二)
 2022-02-28 15:34:03
2022-02-28 15:34:03
来源/作者:普拉特泽-生物医学整体课题外包平台
译:RNF 125和Cbl-b对NLRP 3的连续泛素化抑制炎症小体活化和缓解内毒素血症
JOURNAL OF EXPERIMENTAL MEDICINE( IF :14.307/Q1 )
复制点击此处直达原文:https://doi.org/10.1084/jem.20182091
【内毒素血症】:是由于革兰氏阴性菌细胞壁内的一种脂多糖在细菌死亡后释放入血液,引起发热等一系列的临床综合征。
【NLRP3炎性小体】:核苷酸结合寡聚化结构域样受体蛋白3(NOD-like receptor protein 3,NLRP3)是炎症小体中关键的调控蛋白之一,作为固有免疫的重要组分在机体免疫反应和疾病发生过程中具有重要作用。
【Cbl-b】:Casitas-B系淋巴瘤蛋白-b (Cbl-b)是一种RING finger E3泛素连接酶,在T细胞活化、耐受诱导和分化中起着至关重要的作用。
【RNF125】:一种E3泛素连接酶,作者额外发现该酶参与连续泛素化中。
1、Cbl-b选择性抑制NLRP3炎性小体
由于存在关于Cbl-b参与单核细胞或巨噬细胞中TLR4或myd88介导的信号通路存在相互矛盾的发表观点。因此,作者先确认在TLR配体反应中,Cbl-b是否在促炎细胞因子的产生中发挥重要作用。
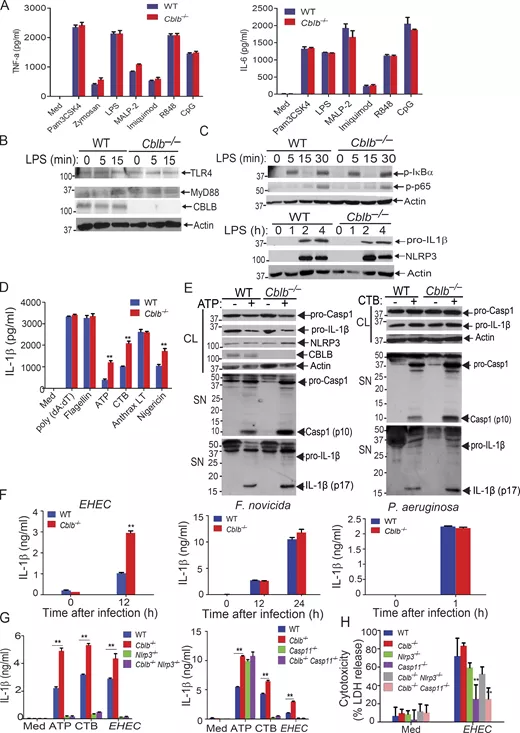
首先用各种TLR配体刺激WT和Cblb - / -骨髓源性巨噬细胞(bmdm),并测量细胞培养上清液中TNF-α和IL-6的生成。没有观察到任何由TLR4或MyD88减少而引起BMDMs缺乏Cbl-b(图1 B)。总之,作者的数据表明,Cblb不调节TLR信号。
为了确定Cbl-b是否调节典型和非典型NLRP3炎症小体,作者用一些能激活典型和非典型炎症小体的毒素来引起敲除小鼠相关症状,检测血液中相关炎症因子。
作者发现WT和Cblb−/−bmdm在炭疽LT、鞭毛蛋白和poly(dA:dT)刺激下产生的IL-1β无差异;然而,显著高于ATP, 尼日利亚菌素,和施莱诱导生产的il-1β,(图1 D)。通过从培养物收集的上清液的免疫印迹显示,在ATP或CTB刺激下,Cblb−/−BMDMs产生的IL-1β增加与活性Casp-1 p10和成熟IL-1β p17的产生增加相关。
为了确定Cblb是否同时调控典型和非典型NLRP3炎症小体,作者将NLRP3缺乏和Casp-11缺乏引入Cblb−/−背景,并生成Cblb−/−NLRP3−/−和Cblb−/−Casp11−/−小鼠。作者通过bmdm测量了IL-1β的产生和在lps -启动和ATP、CTB或EHEC刺激下从WT、Cblb−/−、Nlrp3−/−、Cblb−/−Nlrp3−/−、Casp11−/−和Cblb−/−Casp11−/−小鼠中产生的热死作用。在ATP和CTB刺激或肠出血性大肠杆菌感染时,NLRP3缺乏可消除Cblb−/−bmdm产生的超il -1β(图1g,左图)。相反,Casp-11的缺失只减少了CTB和肠出血性大肠杆菌诱导的超il -1β的产生,而Cblb−/−bmdm诱导的ATP没有产生(图1 G,右图)。肠出血性大肠杆菌诱导的焦亡在WT和Cblb−/−bmdm之间具有可同性,Casp-11缺乏而NLRP3缺乏可显著抑制焦亡(图1 H)。作者的数据支持这样一种观点,即在典型和非典型炎性小体刺激下,Cbl-b调节Casp-11 -和nlrp3依赖的il -1 β的产生,但似乎没有调节EHEC诱导的nlrp3不依赖但Casp-11依赖的自噬过程。(也就是找到了Cbl-b的选择性调节)
为了确定在人巨噬细胞中,Cbl-b是否对NLRP3炎性小体激活有类似的作用,作者采用人单核细胞来源的巨噬细胞进行了实验,,并用cbl-b特异性siRNA转染MDMs或通过核感染扰乱siRNA。在bmdm中敲除CBLB导致在LPS启动和ATP刺激下IL-1β产生升高(图S1),这些结果表明,Cbl-b在小鼠巨噬细胞NLRP3炎性小体中的作用可转译到人巨噬细胞。(效果在人源和鼠源细胞中都得到了印证)这些发现还表明,
Cbl-b可调节由Casp-1和Casp-11分别介导的典型和非典型NLRP3炎症小体。
2、Cbl-b通过Casp-11/ nlrp3依赖的方式抑制lps诱导的亚致死剂量的内毒素血症
LPS的致死性是由髓系细胞分泌的促炎细胞因子IL-1β介导的,或也可在Casp-11和在非常高的剂量下独立于TLR4作用。为了测试Cblb是否在体内调节NLRP3炎症小体,作者用LPS亚致死剂量(5 mg/kg:B4大肠杆菌;通过腹腔注射)。虽然LPS注射后WT小鼠都存活了24小时,但所有Cblb−/−小鼠都死亡了(图2a)。Cblb−/−小鼠的血清TNF-α在2小时显著高于WT小鼠,而IL-1β在注射后2 - 6小时升高。而在WT和cblb−/−小鼠中,血清IL-6水平没有差异(图2b)。为了确定cblb在多微生物脓毒症中的作用,使用盲肠结扎和穿刺(CLP)诱导的脓毒症模型。发现Cblb−/−小鼠对亚致死CLP高度敏感,这与CLP后血液细菌负荷和血清IL-1β升高有关,但与IL-6无关(图2,C和D)。为了确定在Cblb−/−小鼠中观察到的白细胞介素-1βIL-1β升高是否是LPS致小鼠死亡的主要原因,作者在LPS刺激前给Cblb−/−小鼠注射IL-1R拮抗剂(IL-1RA)。如图2a所示,阻断IL-1R可以防止LPS注射后Cblb−/−小鼠的死亡。这种预防与血清IL-1β和TNF-α水平降低有关,但与IL-6水平无关(图2b),这表明IL-1β和TNF-α之间存在正反馈,IL-1β和TNF-α都提高Cblb−/−小鼠的致死率。为了支持这一观点,作者使用了Cblb−/−Nlrp3−/−动物。NLRP3缺陷完全挽救了cblb−/−小鼠,使其免于由LPS亚致死剂量引起的死亡(图2g),这与血清中IL-1β和TNF-α的显著减少有关(图2h)。因此,作者的数据表明,cblb抑制在体内产生nlrp3依赖的IL-1β,以及在LPS刺激下Cblb - / -小鼠血清中TNF-α的增加可能是继发于IL-1β。(IL-1β和TNF-α正反馈,他们的提高可能是LPS诱导的内毒素血症的主要原因)
为了排除适应性免疫系统对脂多糖诱导的Cblb−/−小鼠致死的潜在混淆效应,作者制备了Cblb−/−Rag1−/−小鼠,该小鼠不含T细胞和B细胞。将亚致死剂量的LPS注射到Rag1−/−Cblb−/−小鼠中,再现了在Cblb−/−小鼠中观察到的现象(图S2 a),表明对典型亚致死剂量LPS的致死主要是由固有免疫细胞产生的IL-1β介导的。为了进一步证实这一点,作者制备了带有Cblb等位基因(Cblbf/f;图B S2),缺乏Cbl-b骨髓细胞谱系的交叉LysM Cre老鼠(LysM Cre-Cblbf / f);确实,亚致死剂量的LPS诱导LysM crec - cblbf /fmice的高死亡率(图S2 C),从而再现了在Cblb−/−小鼠中观察到的现象。在表达E3泛素连接酶死亡突变的小鼠中也发现了类似的结果(C373A;2 I),提示Cbl-b对lps诱导的内毒素血症的抑制依赖于其E3泛素连接酶活性。综上所述,作者的数据表明,髓系细胞中Cbl-b的E3泛素连接酶活性抑制NLRP3炎性小体介导的内毒素血症。(找到Cbl-b是泛素化作用来抑制内毒素血症的)
此前的研究表明,lps诱导的致死剂量致死率依赖于Casp-11诱导的焦亡,而不是Casp-1。为了确定Cblb是否也能调节致死剂量LPS诱导的致命性,作者给WT、Cblb−/−、Casp11−/−、Nlrp3−/−、Cblb−/−Nlrp3−/−和一个dCblb−/−Casp11−/−小鼠注射致死剂量(54 mg/kg) LPS。WT, Cblb−−,Nlrp3−−,andCblb−−/ Nlrp3−−/老鼠死后14小时内注射以相似的速度(图2 J,左面板),Casp11 /−−和Cblb /−−Casp11−−/小鼠从注入第一个16 h后存活(图2 J)。注射LPS 20毫克/公斤,WT、Cblb−−,Casp11−−、Nlrp3−−,Cblb−−/ Casp11−−、N d Cblb−−/ Nlrp3−−/老鼠显示缺乏Nlrp3和Casp-11的血清中IL-1β水平的Cblb−−/老鼠(图2 K)。总的来说,这些数据表明,Cbl-b调节Casp-11 / NLRP3炎症小体激活,它触发IL-1β的产生,但它不调节nlrp3独立,但casp -11依赖的非典型炎症小体激活,主要引发焦亡。(致死量救不活了,那就研究亚致死量吧~)
3、在NLRP3炎性小体激活的巨噬细胞中,Cbl-b与NLRP3相关
最近的研究表明NLRP3炎症小体受蛋白质泛素化依赖机制的调控,但泛素化NLRP3的E3泛素连接酶(s)尚未被完全描述。以上数据表明,Cbl-b对NLRP3炎性小体具有负向抑制作用。为了确定Cbl-b是否泛素化NLRP3,作者首先确定Cbl-b是否物理上与NLRP3 相连接。作者通过瞬时转染在HEK293T细胞中表达血凝素(HA)标记的Cbl-b和flag标记的NLRP3。HEK293T细胞缺乏P2X受体,吞噬能力较低,使其对一些nlrp3激活刺激(如ATP和结晶物质,如尿酸单钠和明胶)相对抵抗。因此,尼日利亚红蛋白作为一种离子载体,通过脂质双分子层催化电中性钾/质子交换,诱导NLRP3激活。免疫沉淀flag标记的NLRP3,然后用抗ha抗体进行WB,表明在HEK293T细胞中,Cbl-b与NLRP3相互作用(图3 A)。为了确定Cbl-b - NLRP3关联是否需要ASC,作者使用RAW264.7细胞进行共免疫沉淀(Co-IP)检测。
这里说明一下,凋亡相关斑点样蛋白(ASC)是炎性小体中连接胞浆内受体和半胱天冬酶-1的接头蛋白,在炎性小体活化中ASC聚集成大分子的二聚体,被称为ASC斑点(ASC-speck)。ASC斑点的形成对半胱天冬酶-1的活化至关重要,调控ASC斑点的形成是炎性小体相关疾病的治疗和预防的新途径。
在LPS/ATP刺激下,缺乏ASC的小鼠巨噬细胞Cbl-b与NLRP3相关(图3b),表明它们的关联与ASC无关。为了验证Cbl-b与内源性NLRP3相互作用,作者使用来自C57BL/6小鼠的bmdm进行了Co-IP。同样,在LPS启动和ATP刺激下,Cbl-b与NLRP3结合(图3c)
(此番大费周章就是为了判断Cbl-b与NLRP3泛素化连接,没有之一)
为了确定NLRP3的哪个结构域与Cbl-b相互作用,作者用flag标记的NLRP3及其突变体转染HEK293T细胞(图3 D,上图)。如图3中、下图所示,NLRP3与Cbl-b的相互作用依赖于NLRP3的LRR结构域。最后,作者研究了Cbl-b的哪个结构域与NLRP3的LRR结构域结合。通过使用如图所示的一系列Cbl-b截断片段。3e,作者发现Cblb的UBA区域的缺失使Cblb与NLRP3的结合失效,表明Cblb的UBA区域与NLRP3的LRR区域结合。为了确认Cbl-b UBA区域是否直接与NLRP3的LRR结构域结合,作者将flag标记的NLRP3 LRR或NLRP3ΔLRR与ha标记的Cbl-b UBA一起转染HEK293T细胞。确实,Cbl-b UBA区域直接与NLRP3 LRR结构域结合(图3f)。为了确定Cbl-b UBA是否与NLRP3 LRR结构域上的泛素链结合,作者对LRR结构域内的所有11个赖氨酸残基进行了突变。作者发现NLRP3 LRR K/R突变体未能与Cbl-b结合(图3g),因此支持了Cbl-b UBA区域与NLRP3 LRR结构域上的泛素链结合的模型。用Nlrp3 LRR K/R突变体重组Nlrp3−/−bmdm导致IL-1β产量增加,这进一步支持了这一观点(图3h)。(通过位点突变的方法,找到Cblb的UBA区域与NLRP3的LRR区域直接连接的证据~)
4、Cbl-b以NLRP3为靶点进行泛素化和蛋白酶体介导的降解
为了评估Cbl-b是否针对NLRP3进行泛素化,作者用flag标记的NLRP3、his标记的泛素以及ha标记的缺乏E3泛素连接酶活性的Cbl-b或Cbl-b C373A突变体转染HEK293T细胞。确实,Cbl-b而不是Cbl-b C373A增强了NLRP3泛素化(图4 A和图S3 A)。为了进一步证实Cbl-b是NLRP3的E3泛素连接酶,WT和Cblb C373A BMDMs被LPS激活并被ATP刺激。在来自CblbC373A小鼠的bmdm中,NLRP3泛素化显著降低(图4 B和图S3 B)。这些数据表明,cblb通过泛素化NLRP3抑制NLRP3炎症小体。接下来,作者使用K48和k63特异性泛素抗体确定NLRP3是否经历K48或k63连接的多聚泛素化。事实上,WT bmdm的NLRP3同时经历了K48和k63相关的多聚泛素化。令人惊讶的是,尽管在表达Cbl-b C373A的bmdm中NLRP3的K48链接泛素化减弱,但NLRP3的k63链接泛素化仍未改变(图4 B和图S3 B)。为了证实这一发现,作者用ha标记的Cbl-b和flag标记的NLRP3,以及his标记的泛素,K48泛素,或K63泛素。与之前的一份报告一致,NLRP3接受K48和K63-连接多次泛素化(图4 C和S3 C)。作者的数据表明,Cbl-b介导K48连接的多次泛素化,但K63连接的NLRP3和泛素化的NLRP3 HEK293T细胞中观察到CblbC373A BMDMs可能是由于额外的内生E3泛素连接酶。(这里发现可能有个额外的泛素化连接酶,介导了Cbl-b和NLRP3的连接,因此引出下一章)为了确定NLRP3泛素化的功能意义,作者将WT bmdm以LPS为基础,用ATP刺激或感染EHEC,在存在或不存在MG-132(蛋白酶体抑制剂)或E-64(溶酶体抑制剂)的不同时间点刺激。NLRP3在ATP刺激或EHEC感染时发生降解,MG-132可阻止NLRP3降解,E-64则不能(图4 D)。这些结果表明NLRP3的降解依赖于蛋白酶体。这些数据支持了Cbl-b是NLRP3的E3泛素连接酶并介导NLRP3降解的观点。
5、鉴定 RNF125 作为一种新型 E3 泛素连接酶,诱导 K63 连接的 NLRP3 LRR 结构域的泛素化,该结构域招募 Cbl-b
泛素链与NLRP3 LRR结构域的连接以及它们与Cbl-b UBA区域的关联表明,除了Cbl-b之外,E3泛素连接酶还参与了NLRP3 k63连接的泛素化起始过程。为了识别引发NLRP3 LRR泛素化的E3泛素连接酶,作者进行了GST-NLRP3拉下分析和质谱分析。用LPS刺激WT bmdm 4 h,用ATP刺激5min。用LPS刺激但没有ATP刺激的bmdm作为对照。将BMDM裂解物与GST-NLRP3固定在谷胱甘肽琼脂糖珠上孵育。用SDSPAGE溶解从微珠中洗脱出来的配合物,并通过银染色进行分析(图S4 A)。从微珠中洗脱出来的蛋白被装载在悬浮捕获过滤器上,作为一个细小的分散体被胰蛋白酶消化,通过液相色谱-质谱联用(LCMS/MS)对产生的多肽进行分析,鉴定nlrp3相互作用蛋白。选择潜在的E3泛素连接酶是基于LPS引物或LPS引物+ ATP刺激结合NLRP3,概率为>70%。除了Cbl-b,作者确定了几个额外的E3泛素连接酶包括TRIM213 TRIM21, TRIM47,RNF125和RNF213也绑定NLRP3(图B S4)。注意在这些候选人中,只有Cbl-b E3泛素连接酶必将NLRP3在ATP刺激(图B S4)。
进一步验证这些E3泛素连接酶与NLRP3,他们扮演的角色在NLRP3 炎症小体激活,Co-IP执行实验来关注上述E3泛素连接酶。这些实验表明,在LPS启动和ATP刺激下,TRIM14、TRIM21、TRIM47、RNF213和RNF125在WT bmdm中与NLRP3结合(图5a)。作者沉默TRIM14 TRIM21, TRIM47, TRIM31, RNF213, RNF125 WT BMDMs。发现,沉默RNF125,在LPS启动和ATP刺激下导致IL-1β的产生增加,(找到RNF125这个家伙)数据表明,RNF125是最初的E3泛素连接酶,在其LRR结构域内诱导NLRP3 k63连接的多聚泛素化。作者还注意到HEK293T细胞表达可检测到的RNF125(图5c),这可能解释了为什么NLRP3在不转染外源性RNF125的情况下也能发生K63-和k48 -连锁的多聚泛素化。
为了进一步确定RNF125是否是E3泛素连接酶,诱导k63连接NLRP3在其LRR结构域内的多聚泛素化,作者通过RNF125 siRNA敲除HEK293T细胞中的内源性RNF125,然后用myc标记的RNF125或RNF125环突变体(RM)转染这些细胞。其中包含RNF125环指结构域的C37R和A40M突变,以及在K63泛素存在下flag标记的NLRP3或NLRP3 LRR K/R突变。(RNF125可诱导NLRP3 k63连锁的多聚泛素化),而这种泛素化在表达RNF125 RM或NLRP3 LRR K/R突变的HEK293T细胞中被取消(图5C)。为了进一步证实这些数据,作者用Rnf125 siRNA沉默WT bmdm中的Rnf125基因,用LPS刺激它们,再用ATP刺激。沉默Rnf125废除K63连接多次泛素化NLRP3(图5 D和图S5 B)来验证这个结果,采用K63-特异性去泛素化酶AMSH (SH3域相关分子的信号转导衔接分子),它已被证明特别删除K63泛素链。作者共转染了ha -标记的Cbl-b、flag -标记的NLRP3、myc -标记的RNF125和his -标记的泛素。细胞裂解液用anti-Flag免疫沉淀,然后用或不用k63特异性去泛素化酶AMSH处理。如图5 E所示,用AMSH处理Flag免疫沉淀可以消除K63-而不是k48连接NLRP3的多聚泛素化。这些数据表明,RNF125是E3泛素连接酶的起始位点,通过引导k63连接NLRP3 LRR结构域的多聚泛素化。作者发现RNF125氨基1 - 76片段与NLRP3(图5 F)。自NLRP3经历K63和K48连接多次泛素化,假设RNF125目标NLRP3远程结合域K63连接多次泛素化,它连接Cbl-b NLRP3域。为了验证这一点,用RNF125或RNF125 RM、HAtagged Cbl-b或Cbl-b UBA、flag标记的NLRP3或NLRP3 LRR K/R突变体和he标记的K63泛素转染HEK293T细胞。发现K63泛素链绑定Cbl-b联合NLRP3和K48-连接多次泛素化NLRP3(图5 G n d H和图S5 C),进一步支持这一概念,推倒了在WT BMDMs 中Rnf125废除Cbl-b NLRP3之间的互动,而在没有Cbl-b的情况下,Rnf125 - NLRP3的相互作用仍然保持完整(图5,I和J)。此外,沉默Rnf125基因终止了NLRP3的降解(图5 K)。请注意,沉默Rnf125基因导致NLRP3和pro-IL-1β的表达增加(图5 L)。这表明RNF125抑制了启动过程。总之,数据表明NLRP3分别经历了由RNF125和Cbl-b介导的K63和k48连锁多聚泛素化。(实现连续套娃,RNF125是必不可少的)
6、赖氨酸 496 是将 K48 连接的泛素链连接到 NLRP3 的位点
由于NLRP3的LRR结构域经历了k63连接的多聚泛素化,作者推断Cbl-b介导NLRP3的NBD结构域内k48连接的多聚泛素化。为了确认K496是否是NLRP3的泛素化位点,生成NLRP3 K496R突变体。使用了UbPred:确定了NBD域中的其他四个赖氨酸残基与低到中等的可能性(K324、K430 K510), LS-MS / MS分析也检测到K324和K510(图6)。包括K437 NLRP3 NBD域中,尽管UbPred显示了这个赖氨酸残留没有可能。作者制备了NLRP3 K324R、K430R、K437R和K510R突变体,并用ha标记的Cbl-b、flag标记的NLRP3或NLRP3 K/R突变体和Histagged的K48泛素转染HEK293T细胞。NLRP3在K496R位点的突变完全消除了由Cbl-b诱导的NLRP3的k48连锁多聚泛素化作用(图6 B和图S5 D)。用WT NLRP3重组NLRP3−/−bmdm挽救了k48连锁NLRP3的泛素化作用和随后的NLRP3降解,说明不是NLRP3 K496R(图6 C和图S5 E)。因此,作者数据表明Nlrp3 NBD结构域内的赖氨酸496是k48连接的泛素链连接位点。(K496是泛素链连接位点)
由于RNF125是NLRP3 LRR结构域与k63相关的多聚泛素化启动所必需的,它会招募Cbl-b,因此RNF125缺陷的小鼠也可能对脂多糖诱导的内毒素血症高度敏感,正如Cblb−/−小鼠所显示的那样。最近,作者建立了一种在WT小鼠体内沉默Cblb基因的方案。为了验证这一点,通过尾静脉注射Rnf125特异性siRNA在体内传递来沉默Rnf125基因。与预期一样,在注射LPS亚致死剂量后,所有接受Rnf125 siRNA的小鼠都在40小时内死亡,而接受对照siRNA的小鼠存活率为60%(图7a)。这些数据表明,Rnf125基因沉默小鼠的表型与在Cblb−/−小鼠中观察到的表型相似。(用动物实验进一步验证RNF125的重要性)


电话:400-691-6686
在线时间:8:00-24:00

微信关注公众号


 湘公网安备
湘公网安备